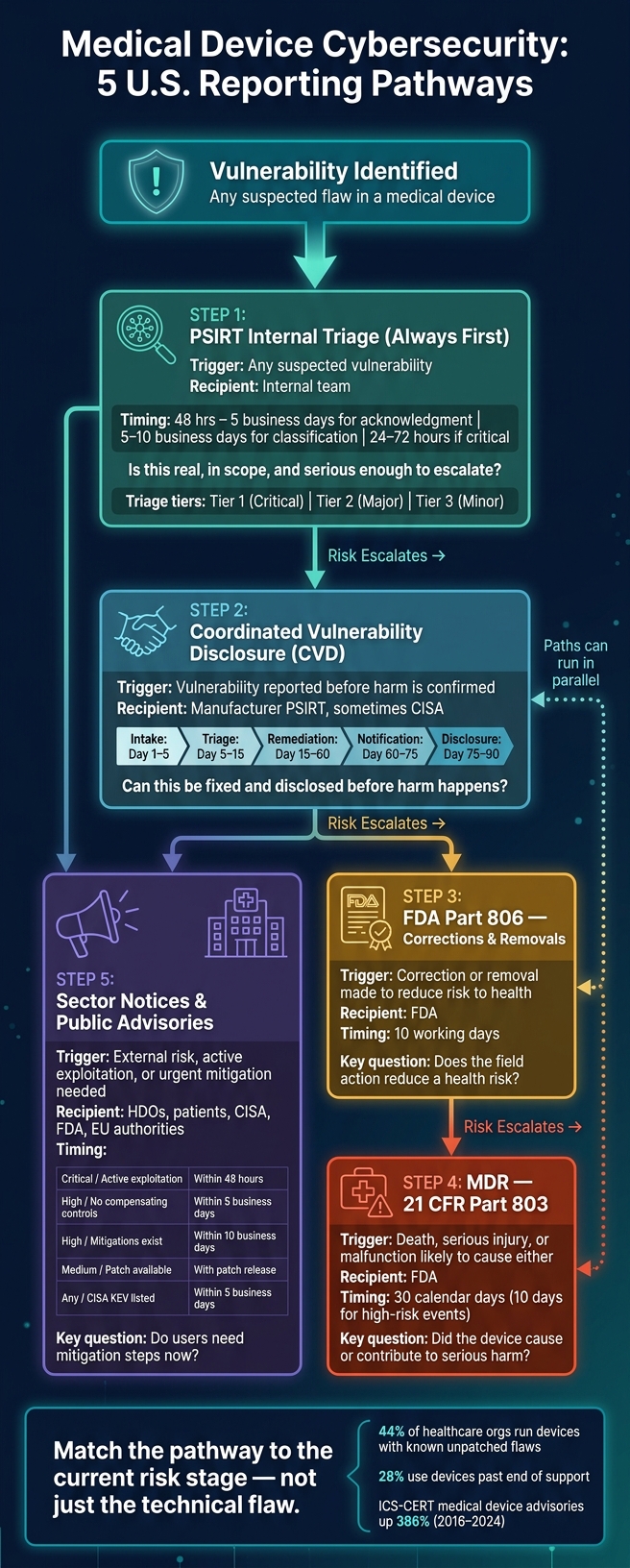
If you route a device vulnerability to the wrong reporting path, you can delay fixes and add patient risk. In this article, I break down the 5 U.S. pathways that matter: PSIRT triage, CVD, FDA post-market reporting under Part 806, MDR under Part 803, and sector/public advisories.
Here’s the short version: PSIRT is the first stop, CVD handles intake and coordinated disclosure, Part 806 covers corrections or removals tied to risk to health, MDR applies when death or serious injury is involved, and public advisories warn hospitals and patients when action is needed fast. That matters more now because 44% of healthcare organizations still run devices with known unpatched flaws, and 28% use devices past end of support.
If I had to reduce the whole article to a simple checklist, it would be this:
- Start with PSIRT for intake, validation, and scoring
- Use CVD when a vulnerability is reported before harm is shown
- Use 21 CFR Part 806 when a correction or removal is made to reduce a health risk
- Use 21 CFR Part 803 (MDR) when a device caused or contributed to death or serious injury
- Use sector notices and public advisories when hospitals, patients, or CISA need timely mitigation guidance
The main rule is simple: match the pathway to the current risk stage, not just the technical flaw.
Medical Device Cybersecurity: 5 U.S. Reporting Pathways Explained
A Quick Primer on FDA's Final Guidance for Cybersecurity in Medical Devices

sbb-itb-535baee
Quick Comparison
| Pathway | Main trigger | Primary recipient | Usual timing | Main patient-safety question |
|---|---|---|---|---|
| PSIRT internal triage | Any suspected vulnerability | Internal team | 48 hours to 5 business days for acknowledgment | Is this issue real, in scope, and serious enough to escalate? |
| CVD | A reported vulnerability needing coordination | Manufacturer PSIRT, sometimes CISA | Often up to 90 days for coordinated disclosure | Can this be fixed and disclosed before harm happens? |
| FDA Part 806 | Correction/removal to reduce risk to health | FDA | 10 working days | Does the field action reduce a health risk? |
| MDR / Part 803 | Death or serious injury, or a malfunction that could do so if it happens again | FDA | 30 calendar days in most cases | Did the device cause or contribute to serious harm? |
| Sector/public advisory | External risk, active exploitation, or urgent need for mitigation | HDOs, patients, CISA, sometimes FDA | 48 hours to 10 business days, based on severity | Do users need mitigation steps now? |
Put another way: these paths are often sequential, not either/or. A single vulnerability can move from PSIRT to CVD, then to FDA reporting and public notice as risk grows.
1. FDA Post-Market Cybersecurity Reporting
Reporting Trigger
FDA post-market reporting starts when a vulnerability creates an unreasonable risk of substantial harm, or when the manufacturer corrects or removes a device to reduce a health risk.
That line matters. Not every vulnerability is reportable to FDA. If a patch is routine and simply tightens security without a direct patient safety impact, it does not need 806 reporting. It still has to be documented through internal tracking and QMS change control [2][4].
FDA Recipient
Once that threshold is met, the next step is figuring out where the report goes.
Reports go to the FDA under 21 CFR Part 806 for corrections and removals, or under 21 CFR Part 803 when the issue caused or contributed to death or serious injury. Put simply:
- Use Part 806 for corrections and removals
- Use Part 803 when the event caused or contributed to death or serious injury [4]
Required Timing
The deadline changes based on the reporting path.
| Pathway | Trigger | Deadline |
|---|---|---|
| 21 CFR Part 806 | Correction/removal to reduce risk to health | 10 working days [4] |
| 21 CFR Part 803 (MDR) | Death or serious injury caused by device | 30 calendar days (10 days for specific high-risk events) [2] |
Risk Threshold
The reporting call depends on documented risk, not just technical severity.
That means manufacturers look at the issue through ISO 14971 and CVSS v4.0 scoring, which adds a Safety metric [2]. A high technical score by itself isn't enough. The key point is whether the risk crosses the FDA reporting threshold.
If a vulnerability falls below that threshold, the reason should be documented in the risk management file. Under QMSR, that rationale belongs in the quality system and needs to stay ready for inspection [1][2][4].
If the issue does not meet this threshold, the next move is usually coordinated vulnerability disclosure.
2. Coordinated Vulnerability Disclosure (CVD)
CVD is the process for receiving, triaging, and disclosing vulnerabilities in a controlled way. Unlike FDA correction or recall reporting, CVD starts before harm is proven.
Reporting Trigger
CVD starts when a vulnerability report comes in through a security email, web form, security questionnaire, or a third-party coordinator like CISA. That report may come from an external security researcher, a customer, or an internal team member [2][6]. For cyber devices, CVD is a required reporting process, not just a nice-to-have. Once a report enters CVD, the next step is simple: figure out who owns triage and response.
Primary Triage Owner
That job usually sits with the manufacturer's PSIRT. PSIRT turns incoming vulnerability reports into a managed disclosure workflow and handles intake, triage, and escalation [2]. If a shared component affects more than one manufacturer, CISA may coordinate disclosure timing across the affected parties [2].
Timing and Disclosure Window
The table below shows the standard CVD phases and the usual timeline for each one.
| Phase | Action | Timeline |
|---|---|---|
| Intake | Acknowledge receipt and assign ID | Day 1–5 |
| Triage | Reproduce vulnerability; develop remediation plan | Day 5–15 |
| Remediation | Build and test the fix; update the SBOM | Day 15–60 |
| Notification | Prepare advisory; coordinate with reporter | Day 60–75 |
| Disclosure | Publish advisory and deploy fix | Day 75–90 |
Source: [2]
These dates can shift based on the quality of the evidence and the likely clinical impact.
Evidence and Patient Risk Threshold
CVD needs enough technical evidence to judge impact. That usually means reproducibility, affected versions, and the clinical use conditions. From there, manufacturers set priority based on exploitability and clinical risk using CVSS v4.0 and ISO 14971 [2]. In plain terms, the issue can move through the security process without waiting for a reportable injury event.
Timely disclosure can cut attacker dwell time and give clinicians earlier notice [3]. A clear CVD policy, along with safe harbor language, can also help protect good-faith reporters [2][6].
3. Medical Device Reporting (MDR) and Recall Pathways
When triage points to likely patient harm, the issue moves out of the security workflow and into FDA reporting. At that stage, MDR and 21 CFR Part 806 become the main escalation paths after PSIRT and CVD have marked the issue as a safety concern.
Reporting Trigger
MDR under 21 CFR Part 803 applies when the facts reasonably suggest that the device caused or contributed to a death or serious injury, or could cause the same kind of harm again if the malfunction happens again [2][4].
Part 806 applies when the manufacturer starts a correction or removal to reduce a risk to health. That can include a software patch [1][4]. In plain terms, if a cybersecurity field action looks and acts like a recall, treat it as a Part 806 correction or removal.
Primary Recipient
This is where things change from CVD. In both pathways, the report goes to the FDA [1][4].
Timing Expectation
Once the case shifts into FDA reporting, the deadline changes too.
| Pathway | Regulation | Timeline |
|---|---|---|
| MDR | 21 CFR Part 803 | Within 30 calendar days of awareness [2][4] |
| Correction or Removal | 21 CFR Part 806 | Within 10 working days of initiating the action [1][4] |
Evidence and Patient Risk Threshold
The main test is simple: does the vulnerability create an unreasonable risk of substantial harm? To answer that, look at both exploitability and the kind of patient harm that could follow [1]. This requires measuring what matters for cybersecurity to ensure clinical continuity.
Examples of harm include:
- Delayed treatment
- Dosing errors
- Loss of monitoring [5]
Routine patches still stay under QMS control, even when they do not trigger MDR or Part 806 reporting [2][1][4].
4. Manufacturer PSIRT Internal Triage
Once CVD intake opens a case, the manufacturer’s Product Security Incident Response Team, or PSIRT, takes over and makes the first triage call.
PSIRT is the first filter. It’s also the lowest-bar path in this process. A case can open for any suspected vulnerability, even if no one has yet shown a safety impact. The point is simple: check the issue early, before anyone knows how serious it is.
Reporting Trigger
A PSIRT case can start from several places, including:
- a dedicated security email
- a structured web form
- bug bounty platforms
- internal penetration testing or code reviews
- threat intelligence feeds from ISAOs or CISA
- direct reports from Healthcare Delivery Organizations (HDOs) [2][4]
Initial Triage Owner
The PSIRT analyst logs the report, assigns an incident ID, and confirms scope [2]. From there, PSIRT pulls in product security, engineering, quality, regulatory, clinical safety, and communications.
| Role | Primary Responsibility |
|---|---|
| PSIRT Lead | Coordination, escalation decisions, regulatory oversight |
| Vulnerability Analyst | CVSS scoring and SBOM correlation |
| Security Engineer | Reproducibility testing and exploit analysis |
| Regulatory Affairs | MDR notification decisions |
| Quality / Compliance | CAPA records and QMS integration (ISO 13485) |
| Clinical Safety | Patient harm assessment under ISO 14971 |
| Product / Communications | Customer notice and advisory timing |
That triage drives the next step. The issue may stay internal, move up to FDA reporting, or head toward public notice.
Timing Expectation
Manufacturers usually send an acknowledgment within 48 hours to 5 business days [2][6]. Initial classification and triage often finish within 5 to 10 business days [2][6].
If the issue is critical, with active exploitation and a patient safety risk, the timeline gets much tighter: 24–72 hours [2].
Evidence and Patient Risk Threshold
PSIRT triage does not need a “risk to health” finding to open a case. That’s a big difference from FDA reporting. If a potential vulnerability is in scope, PSIRT can open it even when the safety effect is still unclear [6][4].
At this stage, the team checks whether the issue can be reproduced, figures out which software versions are affected, and reviews the SBOM against the NVD and CISA’s Known Exploited Vulnerabilities Catalog [2][1]. PSIRT then uses CVSS v4.0 and ISO 14971 to sort both exploit risk and patient risk [2].
Findings are grouped into:
- Tier 1: critical
- Tier 2: major
- Tier 3: minor [4]
If PSIRT finds patient harm or a field correction, the case moves into MDR or recall reporting.
5. Sector Notifications and Public Advisories
When PSIRT confirms that a vulnerability poses risk outside the company, the next move is broad notice. Sector notifications and public advisories tell the healthcare field what happened, who may be affected, and what action to take.
Reporting Trigger
Public advisories are used when a vulnerability creates external patient risk. In most cases, that means a High or Critical issue after PSIRT triage. The same path also applies if the issue is under active exploitation, has no compensating controls, or has moved past the 90-day CVD window.
ICS-CERT medical device advisories increased by 386% between 2016 and 2024 [7].
Primary Recipient
Advisories go to HDOs, patients, and CISA/ICS-CERT. FDA and EU authorities may also receive them [2][7]. TLP labels should be used to limit redistribution.
Timing Expectation
Speed changes with severity. Here’s the expected timeline:
| PSIRT Severity | Trigger Event | Communication Timeline |
|---|---|---|
| Critical | Active exploitation | Within 48 hours |
| High | No compensating controls | Within 5 business days |
| High | Effective mitigations exist | Within 10 business days |
| Medium | Patch available | With patch release |
| Any | Listed in CISA KEV | Within 5 business days |
Source: [5]
Evidence and Patient Risk Threshold
This path is based on possible exposure and patient impact, not on proof that an injury already happened. Section 524B sets the disclosure framework. The public advisory is how that framework gets put into practice when a vulnerability creates an unreasonable risk of substantial harm [2][5].
The goal is simple: get useful mitigation guidance to HDOs before exploitation spreads. Each advisory should include:
- Affected product names, model numbers, and software versions
- A CVSS v4.0 score with the full vector string
- ISO 14971 risk analysis results
- Interim compensating controls, such as network segmentation
- Validated remediation steps, including update versions [2][3]
Those details shape how fast the field can respond.
Pros and Cons of Each Reporting Pathway
Each pathway fits a different trigger. Pick the wrong one, and you can slow down remediation or leave a compliance gap.
| Pathway | Best Use Case | Key Strength | Major Limitation |
|---|---|---|---|
| CVD | Researcher-found vulnerability | Prevents public disclosure before remediation; builds researcher trust | Relies on reporter cooperation; the timeline can lag active exploitation. |
| FDA Post-Market Cybersecurity Reporting (21 CFR 806) | Risk without confirmed patient harm | Formalizes safety actions and regulatory accountability | Requires reporting within 10 working days when a correction is needed to reduce a risk to health; adds QMS documentation work. |
| MDR / Recall | Active exploitation with confirmed harm, or a safety defect requiring removal or correction | Legally mandated; strongest patient-safety response. | Reactive by design; often triggered after harm has already occurred |
| PSIRT Internal Triage | Manufacturer-discovered defect | Fast internal assessment; centralizes scoring and asset mapping. | Internal only; does not satisfy external notification duties by itself |
| Sector Notifications and Public Advisories | Urgent mitigation across hospitals | Broad reach; rapid hospital awareness. | Can contribute to alert fatigue; advisory volume can reduce HDO responsiveness |
The table gives the basic tradeoff. The details below show where each pathway starts to strain.
When a report comes from outside the manufacturer, CVD is usually the first checkpoint. It makes sense for researcher-reported vulnerabilities because it allows controlled remediation before public disclosure. That breathing room matters. It gives the manufacturer time to verify the issue, fix it, and coordinate messaging without turning the problem into a public scramble.
PSIRT plays the internal gatekeeper role. It verifies the report, scores risk, maps affected assets, and sends the case to the right teams, whether that means regulatory, quality, or communications. In plain terms, PSIRT answers the first hard question: What exactly are we dealing with, and who needs to act now?
Once the issue calls for field action, the focus changes. At that point, it’s less about triage and more about getting the word out fast. Sector advisories and public notices work best when hospitals across many sites need shared mitigation guidance right away. The upside is speed and reach. The downside is familiar to anyone in healthcare: too many alerts can blur together, and response rates can slip.
These pathways are often sequential, not exclusive. One vulnerability can set off more than one pathway, so manufacturers need to route the issue based on risk, audience, and timing.
Conclusion
The comparison leads to one practical rule: match the pathway to the current risk stage. No single protocol fits every case. The right path depends on exploitability, clinical impact, and reporting duty.
PSIRT triage is the first step for every vulnerability: intake, validation, and severity scoring. It acts as the gate that routes cases into CVD, FDA reporting, or public advisories.
For reports from outside researchers, CVD is the default model because it gives teams time to coordinate a fix before disclosure. Once the issue turns operational, the focus shifts from disclosure planning to field mitigation. When a vulnerability creates an unreasonable risk of substantial harm, FDA post-market reporting applies. Confirmed or suspected patient harm triggers MDR. [2] Public advisories fit situations where providers need rapid mitigation guidance. [5]
These protocols work in sequence: PSIRT triage, CVD, FDA reporting, and public advisories come into play as risk rises. The point isn’t picking one protocol and ignoring the rest. It’s routing the same vulnerability through the right sequence at the right time.
FAQs
When is a cyber issue FDA-reportable?
A cyber issue is FDA-reportable in three main cases:
- Under 21 CFR Part 806, you must report corrections or removals within 10 working days if they are made to reduce a risk to health.
- Under 21 CFR Part 803, manufacturers must submit MDRs for malfunctions or cybersecurity issues that caused, or could cause, death or serious injury.
- It’s also reportable if a vulnerability turns into an incident that compromises device function or safety.
Can one vulnerability trigger multiple reporting paths?
Yes. One vulnerability or security patch can set off more than one reporting path because medical device cybersecurity sits under overlapping rules.
For example, a single software patch may need to be reported under FDA 21 CFR Part 806 for risk-to-health corrections. That same patch may also need reporting under 21 CFR Part 803 if it relates to an adverse event or malfunction.
In plain English: one issue can lead to more than one filing. Manufacturers need to review each incident against every rule that may apply, not just the first one that seems to fit.
Who decides whether to use PSIRT, CVD, MDR, or Part 806?
For medical device manufacturers, the PSIRT is usually the team at the center of these calls. The PSIRT Lead keeps people aligned, handles coordination, and moves issues up the chain when needed.
Regulatory Affairs makes the final call on notifications under 21 CFR Part 806 or EU MDR. Those decisions are based on the manufacturer’s internal quality management system and postmarket surveillance processes.
