Checklist for FDA-Compliant Medical Device Incident Response
Post Summary
Internet-connected medical devices face growing cybersecurity threats, putting patient safety at risk. To address these challenges, the FDA now requires manufacturers to implement postmarket cybersecurity measures under Section 524B of the FD&C Act, effective March 29, 2023. This includes creating incident response plans, monitoring vulnerabilities, and ensuring timely updates. Key steps for FDA-compliant incident response include:
- Preparation: Build a response team, create a plan, and maintain a Software Bill of Materials (SBOM).
- Detection: Use continuous monitoring systems to identify vulnerabilities and threats.
- Containment: Isolate affected devices while maintaining critical functions.
- Reporting: Submit required reports to the FDA within specified timelines.
- Recovery: Restore device functionality and conduct root cause analysis.
Proper planning and compliance with FDA guidelines are essential to protect patient safety and meet regulatory requirements.
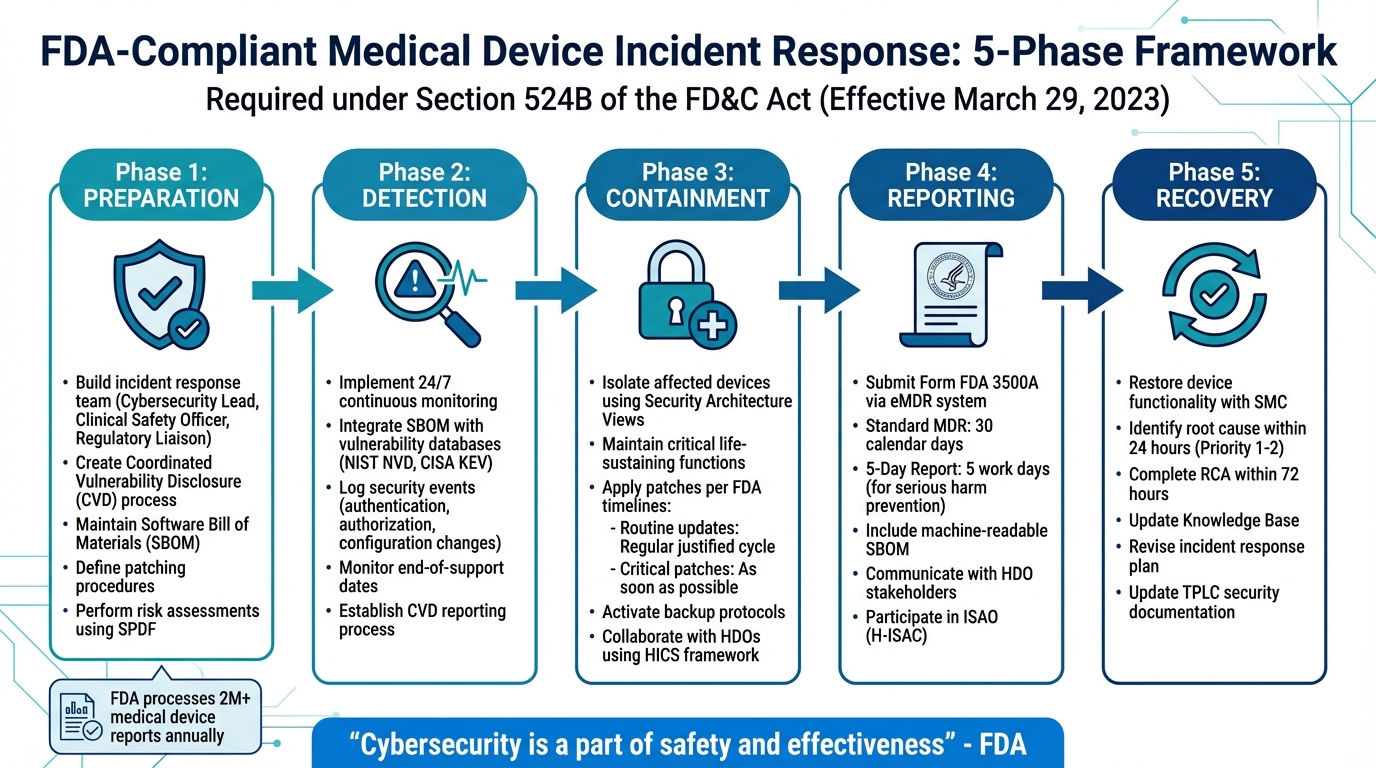
5-Phase FDA-Compliant Medical Device Incident Response Framework
A Quick Primer on FDA's Final Guidance for Cybersecurity in Medical Devices
Preparation Phase: Building Your Incident Response Plan
The FDA now requires manufacturers of "cyber devices" to submit a plan for monitoring, identifying, and addressing postmarket cybersecurity vulnerabilities. This requirement, effective March 29, 2023, under Section 524B of the FD&C Act, is a critical step in ensuring device security and safety [1][6]. This preparation phase lays the groundwork for how your organization will detect, manage, and recover from cybersecurity incidents.
Setting Up Your Incident Response Team
An effective incident response team should bring together specialists in cybersecurity, clinical safety, and regulatory compliance [6]. This mix of expertise ensures that your team can address the full range of security challenges your devices might face. Since cybersecurity is a key component of the Quality Management System Regulation (QMSR), team roles must align with ISO 13485 requirements, particularly for risk management and design controls [6].
Key roles to include:
- Cybersecurity Lead: Oversees technical security measures and threat responses.
- Clinical Safety Officer: Assesses patient safety impacts related to security incidents.
- Regulatory Liaison: Ensures compliance with FDA and other regulatory requirements.
Creating Your Incident Response Plan
Your incident response plan must meet specific FDA expectations, covering several essential elements:
- Coordinated Vulnerability Disclosure (CVD): Develop a clear process for receiving and managing vulnerability reports from external researchers [1][6]. This process should be publicly accessible, with instructions for researchers to contact your organization.
- Patching and Update Procedures: Define how and when updates will be provided. The FDA mandates updates on a "reasonably justified regular cycle" for known vulnerabilities and "as soon as possible" for critical ones [1]. Clearly document what qualifies as a critical vulnerability - typically those posing uncontrolled risks to patient safety - and outline your process for out-of-cycle patches.
- Software Bill of Materials (SBOM): Maintain a machine-readable SBOM to quickly identify affected devices when vulnerabilities arise [1][6].
As Matthew Hazelett, Cybersecurity Policy Analyst at the FDA, emphasizes:
Cybersecurity is a part of safety and effectiveness... A Secure Product Development Framework (SPDF) can be used to fulfill aspects of QS Regulation [1].
Additionally, your plan should include four architecture views: Global System View, Multi-Patient Harm View, Updatability/Patchability View, and Security Use Case View [1][6]. These views are crucial for identifying key system elements, trust boundaries, and potential cascading effects across interconnected devices.
With these elements in place, your plan will provide a structured approach to systematically assess and mitigate risks.
Performing Risk Assessments
Cybersecurity risk assessments for medical devices differ significantly from traditional safety assessments. Instead of relying on historical failure data, these assessments focus on exploitability - how easily a vulnerability can be exploited by an adversary [8][1]. The FDA highlights this distinction:
Cybersecurity risks are difficult to predict, meaning that it is not possible to assess and quantify the likelihood of an incident occurring based on historical data [8].
To address this, use a Secure Product Development Framework (SPDF), such as IEC 81001-5-1, to integrate cybersecurity into the device design process from the start [6]. Your risk assessment should include threat modeling that spans the entire system lifecycle, ensuring traceability between threat scenarios and risk entries [8].
Pay close attention to vulnerabilities listed in CISA's Known Exploited Vulnerabilities (KEV) Catalog. The FDA's guidance is clear:
These vulnerabilities should be designed out of the device, as they are already being exploited [8].
Treat every known vulnerability as a foreseeable risk [8][1], and assume the worst-case exploitability when data is uncertain [8].
Finally, integrate cybersecurity risk assessments with your safety risk management process. Security-related failures that could affect patient safety - such as loss of device functionality or unauthorized access - must be evaluated for clinical impact using ISO 14971 processes [8][1]. These evaluations will directly inform your steps for incident detection and recovery.
Detection and Identification: Finding Cybersecurity Incidents
For medical device manufacturers, spotting problems before they escalate is a top priority. Cyber threats evolve quickly, so identifying vulnerabilities early is essential. As the FDA's postmarket guidance emphasizes:
The exploitation of vulnerabilities may represent a risk to health and typically requires continual maintenance throughout the product life cycle to assure an adequate degree of protection against such exploits [2].
Once your response plan is in place, the focus shifts to continuous detection. Your monitoring systems should run 24/7, scanning for anomalies and matching new threats to your device components.
Setting Up Continuous Monitoring Systems
Your Software Bill of Materials (SBOM) is a key tool for automating vulnerability tracking [1][4]. This inventory lists all software components - commercial, open-source, and off-the-shelf - allowing for automated checks when new threats arise. For example, when vulnerabilities like Apache Log4j surface, an SBOM helps you quickly identify which devices are at risk [3].
Integrate your SBOM with external vulnerability databases for real-time updates. Two critical resources include:
- NIST National Vulnerability Database (NVD): A comprehensive source of known vulnerabilities.
- CISA Known Exploited Vulnerability (KEV) Catalog: Focuses on vulnerabilities actively being exploited - exactly the type of threats the FDA expects you to address immediately [1].
Your devices should also be equipped to detect and log security-relevant events [4]. These might include failed login attempts, unexpected configuration changes, unusual data transfers, or unauthorized network connections. A real-world example: In September 2022, Medtronic identified a vulnerability in its MiniMed 600 Series Insulin Pump System (models 630G and 670G) by monitoring communication protocols. This detection allowed them to issue a correction before patient harm occurred [3].
Additionally, stay on top of end-of-support dates for third-party software [1]. Utilizing automated vendor risk tools can help track these lifecycles across your supply chain. Once a vendor stops supporting a component, your ability to monitor it effectively may diminish, leaving gaps in your defenses.
Defining Incident Criteria and Logging Requirements
Strong monitoring systems are only effective if you can distinguish between minor alerts and serious threats. The FDA suggests categorizing risks as either "Controlled" (acceptable residual risk) or "Uncontrolled" (unacceptable residual risk) [10]. Your incident response should focus on uncontrolled risks - those with the potential to harm patients if exploited.
Logs are another critical piece of the puzzle. They should capture enough detail for forensic analysis but avoid overwhelming your system. Key events to track include:
- Authentication events: Successful and failed logins, as well as privilege escalations.
- Authorization changes: Modifications to user permissions or access controls.
- Configuration changes: Security settings and firmware updates.
- Network activity: Connection attempts and data transfers to unknown IP addresses [4].
Store logs in persistent, non-volatile memory to ensure they cannot be accidentally erased, and limit access to authorized personnel [4]. These logs will be invaluable during FDA inspections or post-incident reviews.
Finally, establish a Coordinated Vulnerability Disclosure (CVD) process to handle reports from external researchers or users [1][4]. These reports often highlight vulnerabilities before they appear in public databases, giving you a critical head start.
Efficiently identifying incidents not only protects patient safety but also helps maintain compliance with FDA requirements.
Containment and Mitigation: Managing Active Incidents
After preparation and detection, the focus shifts to managing active cybersecurity threats. The goal is to contain the threat while ensuring critical functions remain operational. The FDA emphasizes the importance of "availability", meaning any containment strategy must prioritize patient safety above all else [1][12]. Striking this balance is no small task - you need to halt the threat without disrupting life-sustaining device functions.
Isolating Affected Devices
The first step is isolating compromised devices. This process should lean on the device's Security Architecture Views, developed during the design phase. These views map out security domains and external interfaces, enabling you to disconnect affected components without shutting down the entire system [1]. For instance, if a network interface is compromised, you should isolate that connection while the device continues functioning in a safe mode to support critical operations [1][12].
Your Software Bill of Materials (SBOM) becomes invaluable here, helping you quickly identify impacted components. Collaboration with healthcare delivery organizations (HDOs) is also key. Frameworks like the Hospital Incident Command System (HICS) can guide incident management and ensure clinical care continues uninterrupted [7]. Clear, accessible labeling and instructions are crucial, providing HDO staff with the steps to safely disconnect or reconfigure devices while activating backup protocols [1][11].
Once the threat is contained, move immediately to remediation efforts.
Applying Patches and Workarounds
After isolating the issue, remediation is the next step. Under Section 524B of the FD&C Act (effective March 29, 2023), manufacturers are required to provide regular updates for known vulnerabilities and act quickly on critical exploits that pose uncontrolled risks [1][9]. Your Updateability/Patchability View should clearly outline how patches are applied, detailing the system elements and interfaces involved [1].
| Patch Type | Trigger | FDA Recommended Timeline |
|---|---|---|
| Routine Updates | Known unacceptable vulnerabilities | Regular, justified update cycle [1] |
| Critical Patches | Vulnerabilities with uncontrolled risks | As soon as possible (out-of-cycle) [1] |
| Remediation | Postmarket exploits | Within a "reasonable time" [9] |
Every patch must undergo verification through your Secure Product Development Framework (SPDF) to ensure it meets safety and quality standards [11]. In cases where a permanent fix isn’t immediately available, deploy temporary workarounds that have been thoroughly tested to avoid introducing new risks. As the FDA advises:
Instructions to manage cybersecurity risks should be understandable to the intended audience, which might include patients or caregivers with limited technical knowledge [11].
If a workaround requires user action, it must be validated through usability studies to prevent errors that could jeopardize patient safety [1][11]. During this process, activate backup protocols to maintain clinical functionality.
Activating Backup Protocols
To ensure uninterrupted clinical operations, pre-established backup protocols should be activated. The FDA stresses that devices should be designed to operate in degraded states, allowing them to maintain life-sustaining functions even when secondary features, like network connectivity, are disabled [1][12].
Manual protocols for critical functions should be in place to support care during system downtime [7]. For example, if a network-connected infusion pump is compromised, clinicians should have clear procedures for manual dosing and monitoring. The 2017 WannaCry ransomware attack highlighted the consequences of inadequate planning - many healthcare facilities lacked actionable protocols to maintain patient care when digital systems failed [7]. To avoid such scenarios, device labeling should include emergency instructions to guide HDOs in securely reconfiguring or updating devices during an active incident [12].
sbb-itb-535baee
Reporting and Communication: Meeting FDA Requirements
Once remediation efforts are underway, it’s time to focus on reporting and sharing critical details about incidents. The FDA processes over 2 million medical device reports (MDRs) every year, covering suspected device-linked deaths, serious injuries, and malfunctions [5]. These reporting requirements are legally mandated under 21 CFR Part 803 and, for cyber devices, Section 524B of the FD&C Act [5][9].
Submitting Reports to the FDA
Manufacturers are required to report any device-related death, serious injury, or malfunction that could potentially recur [5][13]. Standard adverse event reports must be submitted within 30 days of becoming aware of the issue [13][14]. If immediate action is needed to prevent serious harm, a five-day report is required within 5 business days [13][14].
| Type | Timeline | Trigger |
|---|---|---|
| Standard MDR | 30 Calendar Days | Awareness of device-related death, serious injury, or reportable malfunction [13][14] |
| 5-Day Report | 5 Work Days | When remedial action is needed to prevent serious harm [13][14] |
| Supplemental Report | 1 Month | New information becomes available after the initial report [14] |
All mandatory reports must be submitted electronically using Form FDA 3500A via the eMDR system [5][13]. For cyber devices, manufacturers should also include a machine-readable Software Bill of Materials (SBOM) to help track vulnerabilities [1]. Routine cybersecurity updates addressing "controlled risks" are typically classified as routine enhancements and may not require reporting under 21 CFR Part 806. However, actions to address "uncontrolled risks" that could threaten patient safety must be reported [10].
Coordinating Communication with HDOs
After meeting regulatory reporting obligations, it’s essential to coordinate with healthcare delivery organizations (HDOs) to ensure timely and effective responses. The FDA underscores the importance of collaboration:
Cybersecurity risk management is a shared responsibility among stakeholders including the medical device manufacturer, the user, the Information Technology (IT) system integrator, Health IT developers, and an array of IT vendors [10].
Communication should be tailored to various HDO stakeholders, such as clinical staff, IT administrators, biomedical engineers, and risk managers, as each group needs specific information to act effectively [10][3][14]. For instance:
- Clinical staff: Require details on patient safety impacts and available workarounds.
- IT administrators: Need technical information about vulnerabilities and network changes to implement compensating controls.
The FDA also encourages manufacturers to join an Information Sharing and Analysis Organization (ISAO), like the Health Information Sharing & Analysis Center (H-ISAC), to enhance real-time threat communication. As stated in FDA guidance:
The Agency considers voluntary participation in an ISAO a critical component of a medical device manufacturer's comprehensive proactive approach to management of postmarket cybersecurity threats [10].
Notifications to stakeholders should include a clear explanation of the vulnerability, an assessment of its impact on safety and performance, the potential risk of patient harm, and actionable steps for remediation [10].
Recovery and Post-Incident Review: Learning from Incidents
This phase focuses on restoring normal operations and using the experience to strengthen defenses against medical device security risks. It’s about ensuring devices are safe to use again while capturing key insights to improve cybersecurity practices.
Restoring Device Functionality
Restoring devices to secure and functional operation is a priority before they return to clinical use [7]. The Restoration Team collaborates with the Systems Management Center (SMC) to bring back disrupted services, infrastructure, and applications [15]. It’s essential to document every troubleshooting step, coordination effort, and corrective action in a Post-Incident Report (PIR) [15].
Devices should be built with features that allow for quick and secure updates or patches. Once updates or workarounds are applied, test the device thoroughly to confirm it’s functioning properly and clinical operations can continue safely [7]. Summarize the incident and corrective actions in your organization’s Knowledge Base [15]. This documentation lays the groundwork for a comprehensive root cause analysis.
Conducting Root Cause Analysis
For high-priority incidents (Priority 1 and 2), the FDA requires that root causes be identified within 24 hours of resolving the issue, with a full Root Cause Analysis (RCA) completed and documented within 72 hours [15]. Technical monitors and IT managers must review and approve the PIR and RCA within this timeframe [15].
| Restoration Checklist Item | Requirement/Timeline | Source |
|---|---|---|
| Identify Root Cause | Within 24 hours of resolution | FDA SMG 3210.13 |
| Complete RCA | Within 72 hours of resolution | FDA SMG 3210.13 |
| Knowledge Base Update | Post-resolution | FDA SMG 3210.13 |
| PIR Sign-off | IT Managers/Technical Monitors | FDA SMG 3210.13 |
During the RCA, look for factors that may have worsened, delayed, or concealed the incident [15]. Threat modeling can help identify objectives and vulnerabilities while outlining countermeasures to reduce risks [16]. Assess whether the remaining risk is “controlled” (acceptable) or “uncontrolled” (requiring immediate action) [16].
Your Software Bill of Materials (SBOM) can accelerate the analysis by pinpointing affected areas [1]. If third-party software was involved, update the SBOM and security architecture to reflect any new dependencies or mitigations [12]. These updates not only improve cybersecurity defenses but also shape future risk mitigation strategies.
Improving Your Incident Response Plan
The lessons learned during restoration and root cause analysis should directly influence updates to your incident response plan. The aim is to eliminate high-priority incidents and significantly reduce recurring issues [15].
Post-incident findings should trigger updates to the device’s risk assessment, factoring in the likelihood of exploitation, potential impact on performance, and severity of harm to patients [16]. By aligning risk management documentation with these insights, your plan stays resilient against emerging threats. Use these updates to refine Total Product Lifecycle (TPLC) security risk management documentation and cybersecurity plans [1].
Adopting a Secure Product Development Framework (SPDF) helps manage cybersecurity risks throughout the TPLC - from design and development to support and decommissioning [12]. Keep in mind that cybersecurity measures can weaken over time as new threats surface, making regular updates to the response plan essential [12].
Incorporate incident insights into revised policies, simulations, and staff training to prevent similar events. Assess how well coordination with Healthcare Delivery Organizations (HDOs) worked and ensure communication channels for "cybersecurity signals" are effective [16][7]. Active participation in Information Sharing Analysis Organizations (ISAOs) is also key to staying ahead of postmarket threats and vulnerabilities [16].
Using Censinet RiskOps™ for Medical Device Incident Response
Censinet RiskOps™ takes medical device incident response to the next level by streamlining processes that are often complex and time-sensitive. When dealing with FDA-regulated device incidents, healthcare organizations face challenges like coordinating diverse teams, tracking vulnerabilities in real time, and meeting strict documentation standards. This platform simplifies these tasks by automating workflows, encouraging collaboration, and making regulatory reporting more manageable.
Automating Vulnerability Management
Censinet RiskOps™ simplifies vulnerability management by automating the tracking and prioritization of risks across your medical device inventory. Considering that 53% of connected devices have at least one unpatched critical vulnerability, this feature is crucial [17]. The platform continuously updates its Software Bill of Materials (SBOM), ensuring that component versions and dependencies are current. This makes it easier to quickly assess and address new threats as they arise [17].
The system classifies risks into two categories: "uncontrolled" risks, which demand immediate updates, and "controlled" risks, which can be addressed during scheduled maintenance. This categorization uses tools like the Common Vulnerability Scoring System (CVSS) [17]. Such prioritization aligns with Section 524B of the FD&C Act, effective since March 29, 2023, which requires manufacturers to monitor and address vulnerabilities and exploits after devices are released to market [3][4]. Automated workflows also support coordinated vulnerability disclosure (CVD) policies, ensuring that vulnerabilities are reported within the mandated 30-day timeframe [17].
Facilitating Collaborative Risk Assessment
Censinet RiskOps™ acts as a centralized hub for collaboration during incidents, bringing together healthcare delivery organizations (HDOs), manufacturers, and internal teams. By integrating with existing response strategies, the platform automates the creation and storage of communication logs, which are essential for FDA reporting. This eliminates the need for manual documentation during critical situations.
The platform also supports participation in Information Sharing and Analysis Organizations (ISAOs) like H-ISAC and Sensato. These organizations provide structured environments for sharing information about vulnerabilities and emerging threats with the FDA and other stakeholders [3]. This collaborative approach seamlessly transitions into automated compliance management, ensuring that every incident is documented and reported according to FDA requirements.
Simplifying Compliance and Reporting
Censinet RiskOps™ centralizes all cybersecurity documentation required under federal mandates, making it easier to keep incident response plans, vulnerability disclosures, and risk assessments organized and ready for audits. The platform automates the generation of reports for FDA premarket submissions and postmarket surveillance, reducing manual effort while ensuring compliance with FDA guidance FDA-2021-D-1158.
With real-time risk visualization dashboards, healthcare organizations can demonstrate active monitoring and response to device vulnerabilities, meeting regulatory expectations. The platform integrates these cybersecurity documents into the Quality Management System (QMS), tracking risk management throughout the device lifecycle [4]. Additionally, organizations can export their continuously updated SBOM and vulnerability logs at any time to fulfill FDA documentation requests related to device risk and resilience.
Conclusion
Protecting cybersecurity in medical devices isn’t just about safeguarding technology - it’s about protecting patients. As the FDA puts it, "Cybersecurity is a part of safety and effectiveness" [1]. Since the implementation of Section 524B of the FD&C Act on March 29, 2023, ensuring device security throughout its entire lifecycle has become a regulatory necessity [1][3].
The checklist's five phases - preparation, detection, containment, reporting, and recovery - create a comprehensive framework to address both immediate threats and ongoing security risks. By following this structured approach, organizations can better navigate the complex challenges of securing medical devices.
Starting October 1, 2023, the FDA began issuing "Refuse to Accept" decisions for premarket submissions that fail to include adequate cybersecurity documentation [11]. This emphasizes the importance of treating cybersecurity as a fundamental part of the device lifecycle, rather than an afterthought [1].
FAQs
What qualifies as a “cyber device” under Section 524B?
A "cyber device", as defined in Section 524B of the FD&C Act, refers to a device that meets the following criteria: it includes software that has been validated, installed, or authorized by the sponsor as part of or within the device; it has the capability to connect to the internet; and it contains technological features that could be susceptible to cybersecurity threats.
When does a cybersecurity issue trigger an FDA MDR or 5-day report?
If a cybersecurity issue leads to a device causing or contributing to a death or serious injury - or if a malfunction could potentially result in such outcomes if it happens again - it triggers an FDA Medical Device Reporting (MDR) requirement. In such cases, manufacturers are required to file a report within 30 days, or within 5 working days if the FDA specifies this expedited timeline in its regulations.
How do we keep patient-critical functions running while isolating a compromised device?
To keep essential patient functions running while isolating a compromised device, disconnect it from the network to contain the threat and stop further risks. In the meantime, rely on backup systems or alternative workflows to ensure critical operations remain unaffected. Having clear protocols in place, working closely with device manufacturers, and preparing contingency plans are all crucial steps to safeguard patient safety and maintain smooth operations during the isolation process.

